Examples
This series of examples is meant to give guidance on how to perform a series of tasks with TSCoDe, hoping that some of these examples will be very similar to the task you are trying to automate. Play around!
For detailed descriptions of the operators and keywords present in the inputs, see Operators and keywords.
Work is in progress to expand this section with more examples.
1. Generation of a 3D structure from SMILES, conformational search and refinement
LEVEL=GFN-FF
refine> rsearch> opt> O=C(O)[C@H](NC([C@H](NC([C@@H](C)N)=O)C)=O)C
# This is a comment line!
# First row sets the level of theory at the GFN-FF level via XTB.
# If XTB is not set as the default calculator, you can specify it
# here adding CALC=XTB in the keyword line.
# First, the SMILES string is converted into a 3D structure
# (H2N-Ala-Ala-Ala-OH), then operators are applied starting
# from the inside out:
# opt> - the generated 3D structure will be optimized
# rsearch> - a knowledge-based, torsion-based conformational search
# is carried out. The number of conformers generated can be adjusted
# with CONFS=n (default is 1000)
# refine> - takes the 1000 conformers ensemble and starts refining it
# by discarding identical structures, optimizes it at the GFN-FF level
# and repeats the pruning steps. For details on the pruning, see the
# "How TSCoDe Works" section
1. Trimolecular input
DIST(a=2.135)
maleimide.xyz 0A 5x
opt> HCOOH.xyz 4x 1y
csearch> dienamine.xyz 6A 23y
# First pairing (A) is the C-C reactive distance
# Second and third pairings (x, y) are the
# hydrogen bonds bridging the two partners.
# Fixed constraints (A, UPPERCASE letters) will refine to the imposed values (here a=2.135 A)
# Interaction constraints (x, y, lowercase letters) will relax to an optimal value
# opt> - structure of HCOOH.xyz will be optimized before running TSCoDe
# csearch> - A conformational search will be performed on dienamine.xyz before running TSCoDe

Best transition state arrangement found by TSCoDe for the above trimolecular input, following imposed atom spacings and pairings
2. Atropisomer rotation
SADDLE KCAL=10
scan> atropisomer.xyz 1 2 9 10
# scan> : (four indices specified) performs two dihedral
# scans (clockwise/anticlockwise) rotating the specified
# dihedral angle in 10° increments. Then, peaks above
# 10 kcal/mol (KCAL keyword) form the lowest energy
# structure are re-scanned at increased accuracy (1°
# increments).
# SADDLE: Each maxima is then optimized to a saddle point.
# It is also possible to replace SADDLE with NEB to use scan
# points to run a NEB in an automated way.

Best transition state arrangement found for the above input

Plot of energy as a function of the dihedral angle (part of TSCoDe output).
3. Peptide-substrate binding mode
RMSD=0.3
csearch> hemiacetal.xyz 34x
csearch_hb> peptide.xyz 39x
# Complex binding mode between a reaction
# intermediate (hemiacetal) and the catalyst
# (peptide).
# RMSD=0.3 reduces the similarity threshold to
# retain more structures (default 0.5 or 1 A)
# csearch> performs a complete conformational
# search on hemiacetal.xyz (2 diastereomers,
# total of 72 conformers)
# csearch_hb> performs a partial conformational
# search on peptide.xyz, retaining the hydrogen
# bonds present in the input structure. 19683
# conformers are generated, and the most diverse
# 1000 are used for the embed (override with CONFS=n)
# String algorithm: 5.18 M poses checked

Input structures for hemiacetal.xyz (left) and peptide.xyz (right)

Best pose generated for the above input. The yellow bond is the imposed interaction, dotted lines are hydrogen bonds
4. Complex embedding with internal and external constraints
DIST(a=2.0, x=1.6, y=1.6) SOLVENT=ch2cl2 KCAL=20
mtd_search> quinazolinedione.xyz 6A 14A 0x 7y
csearch> peptide.xyz 0x 88y 19z 80z
# Four pairings provided (A, x, y, z):
# A - Fixed (UPPERCASE letter), internal to quinazolinedione
# (green) - kept at 2.0 Å during the entire run
# x - Interaction (lowercase letter) - will be embedded at 1.6 Å
# and then relaxed during the ensemble optimization steps (red)
# y - Interaction (lowercase letter) - will be embedded at 1.6 Å
# and then relaxed during the ensemble optimization steps (orange)
# z - Interaction (lowercase letter), internal to peptide (light blue)
# No distance provided, will relax during optimization
# mtd_search> - metadynamics-based conformational search through CREST.
# Note that this is internal constraints-aware, and will treat the "A",
# "x" and "y" pairings as bonds, retaining the specified distances.
# csearch> - diversity-based torsional conformational search. As rsearch>,
# it is constraints-aware and will treat the "z" pairing as a bond, preventing
# the generation of peptide conformers without the "z" interaction present.
# The KCAL keyword sets the energy threshold in kcal/mol for both the final
# ensemble and the metadynamics-based conformational search ("--ewin" in CREST).

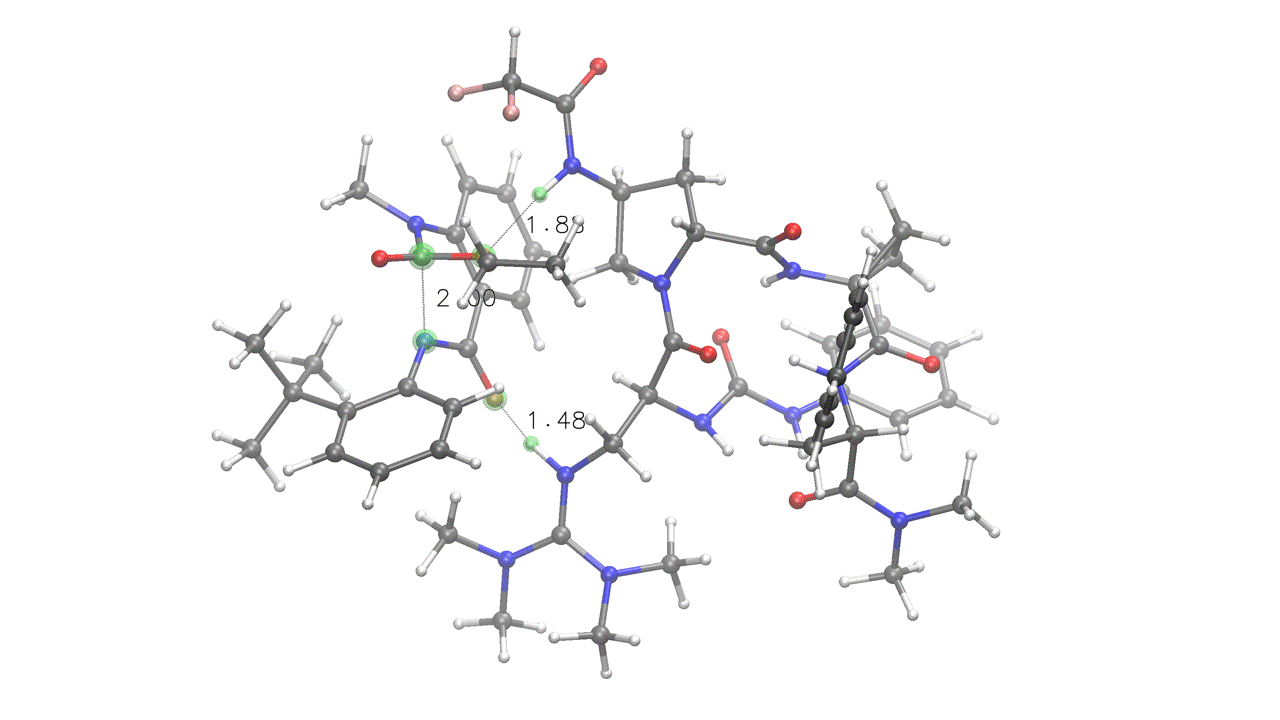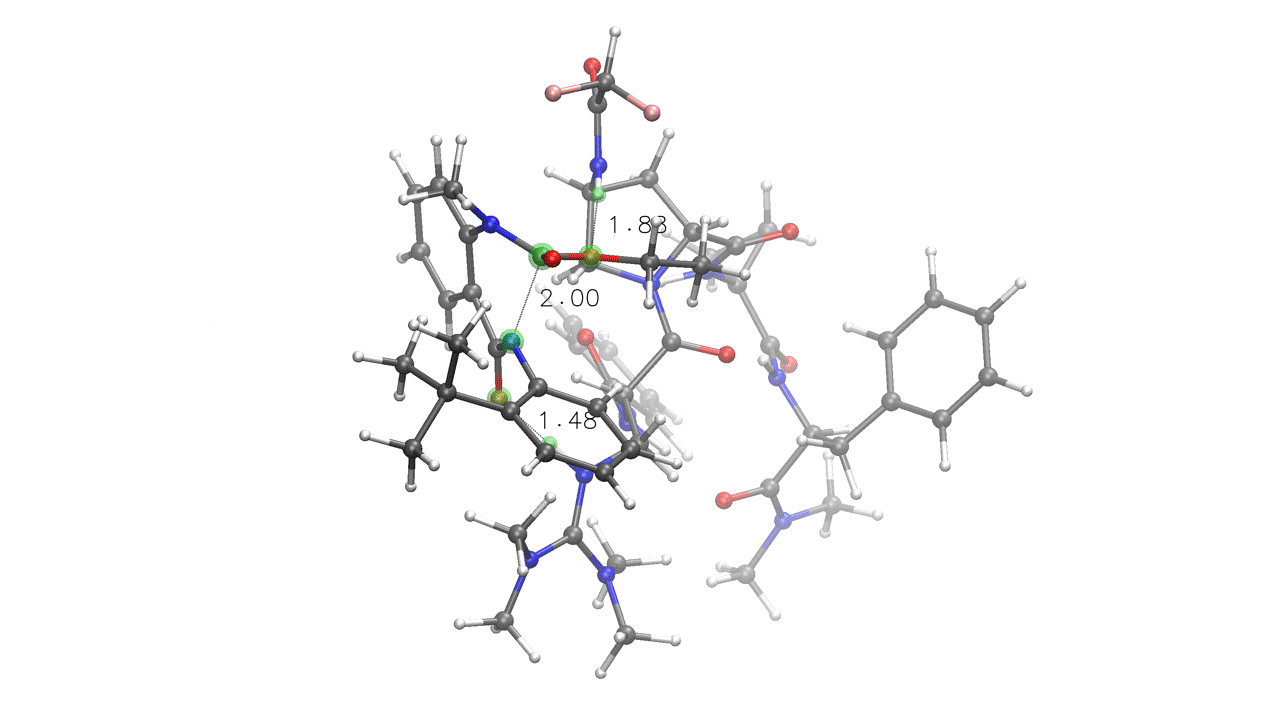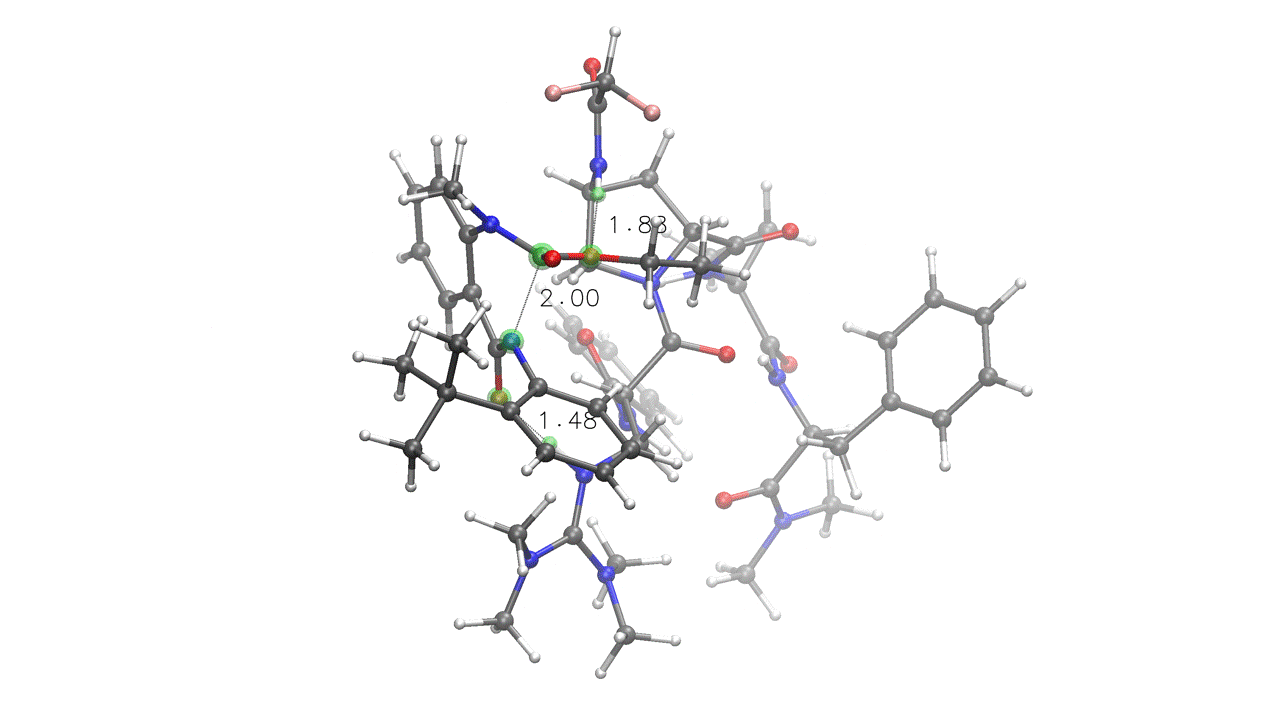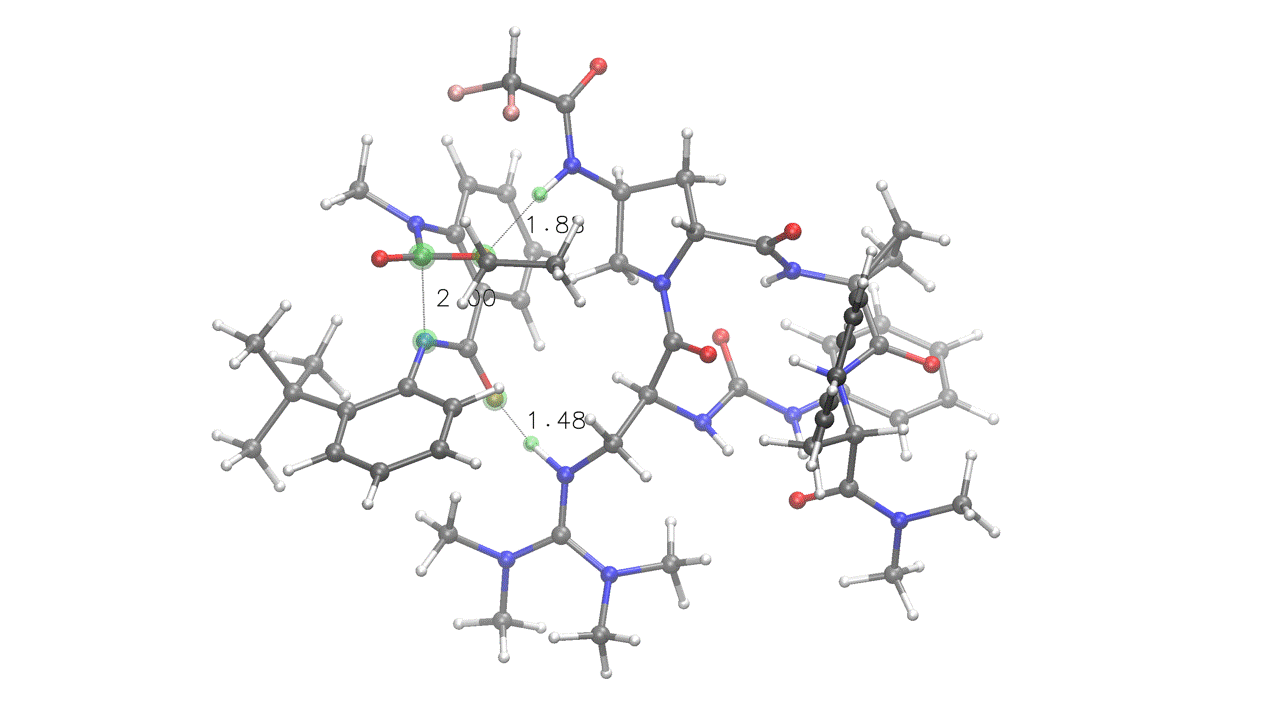
One of the poses generated for the above input. Note how fixed constraints were mantained (a=2) while interactions were relaxed (x=1.6, y=1.6, z)